INTRODUCTIONVogt-Koyanagi-Harada (VKH) syndrome or Uveoencephalitis is a rare systemic disease involving various melanocyte-containing organs, such as eyes, meninges, central nervous system, skin, membranes, mucosas and inner ear (cochlea and vestivular system) (1).
The disease has been reported since 9
th century, when doctor Ali Isa observed the first eye inflammation followed by ciliary dyspigmentation (2).
Scientists such as Schenkl (1873), Vogt (1906), Harada (1926), Koyanagi (1929) and others reported independent cases of patients with uveitis associated with deafness, vitiligo and hair loss. During the first half of 20
th century, previously described disorders were named as an eponymous Vogt-Koyanagi-Harada syndrome (2,3,4,5).
It affects more frequently Africans, Indians and Latin-Americans, as well as affecting individuals with darker pigmentation, especially women (ratio of 2:1). The beginning of the disease usually occurs between second and forth decade of life. Nevertheless, when it happens during childhood, its recurrence is associated to a more aggressive development (3,6).
The routine at the
Hospital Universitário Bettina Ferro de Souza - Universidade Federal do Pará and
Universidade do Estado do Pará in Otorhinolaryngology, Ophthalmology, Neurology and Dermatology areas was the main reason for the accomplishment of this study on Vogt-koyanagi-harada syndrome. This study aims to make health professionals aware of the syndrome occurrences. Although rare its incorrect diagnosis and therapy can lead to severe complications.
The objective study to review the literature on the several aspects of Vogt-koyanagi-harada syndrome, focusing on its pathogenesis as well as its clinical ENT manifestations.
METHODOnline database research provided useful information and articles for this study. They can be accessed at any time for researching and updating, as they are built up according to the progress of scientific literature production.
The databases consulted were Cochrane, LILACS, MEDLINE, OMIM and SciELO, by applying to the research the terms Vogt-koyanagi-harada syndrome and Uveoencephalitis syndrome for articles published between 1997 and 2007, besides others.
PathogenesisThe etiologic and pathogenic factors in VKH syndrome remain unclear, though evidences of an autoimmune process against an antigenic component on the melanocyte. There is an association with Hashimoto's thyroiditis and Rheumatoid Arthritis (7,8,9,10).
It has also been associated with major histocompatibility complex (MHC II); DR4 and with HLA-DRB1*0405 allele. The former is considered the most expressive in Brazilian patients (11,12).
The presence of such allele determines the recognition by the T lymphocytes of peptide derived from tyrosinase and other proteins from the same family that are found on melanocytes, by establishing though a cellular immune inflammatory response, with a predominance of T helper type 1 lymphocytes (13).
It is still unknown whether such process is idiopathic or caused by an infection (14). Some studies invoke a possible role of Epstein-Barr virus reactivation in this disease. Though this needs more data to be established (15,16).
Clinical manifestationsClinical manifestation of Vogt-koyanagi-harada syndrome can be prodromal, uveitic, chronic and of recurrence (17).
In the prodomal stage, patients can present symptoms including headache, low-grade fever, photophobia, nuchal rigidity, weak muscles, hemiparesis, dysarthria and aphasia, as well as lymphocytic pleocytosis, an increase on pressure and protein on liquoric analysis. Patients can present skin and hair sensitiveness. For some period though, patients might not present such symptoms or atypical signs as optical neuritis and paralysis of the peripheral nerve (1,2,18).
The disease develops to uveitic stage presenting symptoms as photophobia, conjunctival hiperemia, loss or blur of vision and ocular pain. This stage can last from weeks to months and it is commonly when patients search for treatment. In clinical terms, bilateral posterior uveitis is associated with retinal oedema, hyperemia and oedema of the optic disc by occasionally displacing the retina (18). It often affects the ocular anterior chamber with keratic precipitates in lamb lard and iris nodules. Intraocular pressure can be high (2). Some patients, in this stage, can present ENT manifestations (19).
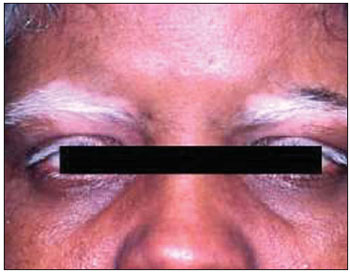
Picture 1. Achromatic stain on the eyebrow (poliosis) - By: Brito AE; Zagui RB; Rivitti EA; Nico MM. Você conhece esta síndrome? An Bras Dermatol. 2005: 80(3):297-8.
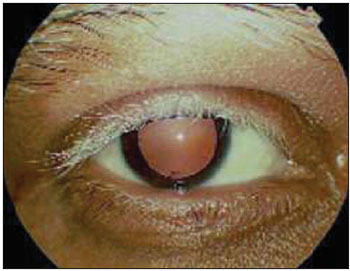
Picture 2. Anterior Uveitis - keratic precipitates - By Brito AE; Zagui RB; Rivitti EA; Nico MM. Você conhece esta síndrome? An Bras Dermatol. 2005: 80(3):297-8.
The chronic stage can last from months up to years. Besides ophthalmological symptoms, cutaneous signs as eyelash, eyebrown and hair poliosis, symmetric vitiligo on face are also signs for diagnosis (1,2,18).
Recurrence stage is characterized by recurrent granulomatous panuveitis combined with complications as cataract and glaucoma (20).
Diagnosing these two last stages is not simple, especially if patient does not report prodomal signs (21).
Otorhinolaryngology manifestationsENT manifestations involve inner ear, cochlea and vestibular system, and can account for 75% of the cases. Symptoms of central origin develop together with the findings of uveitic stage (22).
The physiopathological basis of symptoms lies on inflammation and loss of melanocytes, which are essential to the perfect activity of the inner ear. The production of specific antibody against inner ear proteins is also affected (23,24).
The vestibulo-cochlear nerve is, as the second pair of cranial nerves, the main involved one, by leading to sensorineural hearing loss, which is comprised among the neurological peripheral causes of sudden sensorineural hearing loss (2). Sudden sensorineural hearing losses are the ones of 30 dB or above, in 3 contiguous audiometric frequencies, occurring in three days. Tympanic alterations that might suggest a component of conduction on hearing loss are not noticed (25).
Hypoacusis is, in general terms, bilateral and progressive by damaging the ability of understanding words and acute sounds or all sounds. The type of development of hearing alteration is of an autoimmune-caused hypoacusis. Tinnitus is present in 50% of the cases (26,27).
The involvement of vestibular system is less frequent leading to vertigo, horizontal nistagmus and alteration on vestibulo-ocular reflex. Nistagmus on first gaze position does not occur. The lack of auditory symptoms associated to nistagmus denies syndrome and requires investigation of central causes to the involvement of vestibular system (28).
Kimura and col. studied 20 patients with VKH syndrome and were able to show clinical and laboratory alteration in their inner ear, by making evident the important otoneurological involvement (24).
The presence of ENT manifestations does not influence recurrences or development of the complications. Such manifestations present satisfactory prognosis when there is an improvement coming from suitable therapy (30).
DiagnosisDiagnosis of such a syndrome is achieved by signs and symptoms derived from diagnosis criteria. There is no specific test to confirm it (1).
In 1978, American Uveitis Society recommended the first criteria for possible diagnosis of such syndrome (1), and in 1999 the I International Workshop on VKH Syndrome took place, aiming the review and improvement of the criteria. These concern multi-systemic nature of the disease including ophthalmological conditions at the early and late stage of the disease. Then, a criterion was established according to the amount of sings as complete, incomplete or possible syndrome (4).
As criteria of the disease, they can be: 1) absence of history of penetrating ocular trauma or surgery preceding uveitis; 2) absence of clinical or laboratory history of other type of ocular disease; 3) bilateral ocular involvement; 4) presence of neurological/auditory findings (as they might have disappeared in the moment of clinical presentation) or, 5) cutaneous findings. And these last ones cannot precede either ocular disease or neurological manifestations (4, 31).
Patients with VKH syndrome of complete type should present all clinical criteria properly filled up. Patients with VKH syndrome of incomplete type present ocular involvement filling up the three first criteria combined with the presence of neurological and auditory alterations or cutaneous alterations (32).
Syndrome is considered of possible type when presenting only ophthalmological criteria; terminology such atypical VKH cannot be used (4).
Patients with hearing loss, tinnitus, dizziness and associated manifestations should be submitted to auditory and body balance tests. The selection of tests depends on the clinical history of patients, and audiological tests might help on diagnosis by the great amount of patient involved (28).
For being an autoimmune disease, it is necessary to perform tests as hemosedimentation, hemogram, mucoprotein, rheumatoid factor, circulating immune complexes, anti-nuclear antibodies, anti-collagen antibodies II, total complement and fractions to evaluate inflammatory activity as well as the investigations of other associated disorders (7,8).
The intolerance to glucose should be analyzed, though it may be present in 55% of patients (33).
Differential diagnosisA wide differential diagnosis should be listed in cases when ocular signs and symptoms are combined with neurological and auditory manifestations and cutaneous diseases. VKH syndrome should be considered when all hypothesis of rheumatological, neoplasic and infectious diseases in a immune-competent patient are dismissed. Inflammatory diseases, like differential diagnosis, can be: Systemic Erythematous Lupus, Sarcoidosis and Behçet's disease. Differential diagnosis can be performed with infectious diseases such as Syphilis, Tuberculosis, Herpes, Fungus Infection, Toxoplasmosis, Acquired Immunodeficiency Syndrome AIDS (1,34).
Ophthalmological and Central Nervous System diseases can also be classified as differential diagnosis, which comprise Sympathetic ophthalmia, Posterior Acute Multifocal Epitheliopathy and Multiple displacement of the secondary retina to systemic blood hypertension.
ENT involvement should be done with vestibular neuronitis, Lyme disease and Cogan's syndrome (35).
TherapyTherapy must be early and intense, thus quickly diagnosing is important. After symptoms improve, weaning should slowly occur between 3 and 6 months, and be remained for 1 year if recurrence might occur (36).
The objective of therapy is to suppress inflammation on target tissues of the disease by making use of immunesupressor doses of corticosteroid. The process can be via oral (prednisone 1-2 mg/kg/day) or via pulsotherapy (metilprednisolon 1g/day from 3 to 5 days) (1). Pulsotherapy by intravenous medication has been the one used on Vogt-koyanagi-harada syndrome (18,32).
Other types of corticoids can be used when suspecting of hepatic disorder and tests of abnormal lymphocyte function. They can be Dexametasone (20mg/day) or Betamethasone (30mg/day), presenting the possibility of positive therapy response (36,37).
In patients who present few or no responses, the human Immunoglobulin IV or immunesupressors as ciclosporin (most used), azathioprine, ciclophosfamida, methotrexate can be used for one year at least (38).
The accurate therapy in otoneurology depends on the precise syndromic, topographic and etiological diagnosis. A multiple therapy approach is the best action regarding vertigo and other types of dizziness of vestibular origin. Corticotherapy reduces symptoms (15).
Nowadays, several studies have been developed in order to evaluate the use of Adalimumab, a type of drug acting as an anti-Tumor Necrosis Factor (anti-TNF) on Vogt-koyanagi-harada syndrome (39).
PrognosisThe important action for a positive prognosis is an immediate start of therapy by taking high doses of anti-inflammatory medication in order to control inflammation (40). Hearing function is recovered most of cases, while cutaneous injuries might remain (15).
Visual prognosis is related to the development of glaucoma and subretinal neovascular membrane during chronical stage. Final visual acuity, when better than 20/40, occurs in only 30% of the cases, which means an important cause of blindness (37).
FINAL CONSIDERATIONSAs Vogt-Koyanagi-Harada syndrome is a rare disease, its diagnosis becomes a challenge (1), and it is essential to be early performed. The diagnosis of clinical deafness, tinnitus and dizziness should always consider Vogt-Koyanagi-Harada syndrome. The presence of ophthalmology, otorhinolaryngology, neurology and cutaneous alterations requires an interdisciplinary medical team, by actively participating on different or correlated fields in order to manage therapy and clinical evolution, by improving the patient's prognosis.
BIBLIOGRAPHICAL REFERENCES1. Matiello M, Carvalho HC, Alvarenga H, et al. Síndrome de Vogt-Koyanagi-Harada. Cad Bras Méd. 2004, 17:50-8.
2. Moorthy R, Inomata H, Rao NA. Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol. 1995, 39(4):265-92.
3. Tabbara KF, Chavis PS, Freeman WR. Vogt-Koyanagi-Harada syndrome in children compared to adults. Acta Ophthalmol Scand. 1998, 76:723-6.
4. Read RW, Holland GN, Rao NA. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol. 2001, 131(5):647-52.
5. Belfort R, Nishi M, Hayashi S, et al. Vogt-Koyanagi-Haradas disease in Brazil. Jpn J Ophthalmol. 1988, 32(3):344-7.
6. Mondkar SV, Biswas J, Ganesh SK. Analysis of 87 Cases with Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol. 2000, 44:296-301.
7. Wiesli P, Bernauer W, Furrer J. Headache and bilateral visual loss in a young hypothyroid Indian man. J Endocrinol Invest. 1999, 22:141-3.
8. Shinzato M, Yamamoto J, Hirata CE, et al. Eye disease in a patient with rheumatoid arthritis. Postgrad Med J. 1999, 75:676-7.
9. Ishikawa A, Shiono T, Uchida S. Vogt-Koyanagi-Harada disease in identical twins. Retina. 1994, 14:435-7.
10. Zaoutis LB, Rose CD, McKay CP. Intrafamilial occurrence of tubulointerstitial nephritis with uveitis and Vogt-Koyanagi-Harada syndrome. J Rheumatol. 1999, 26:2496-8.
11. Levinson RD, See RF, Rajalingam R, et al. HLA-DRB1 and -DQB1 alleles in mestizo patients with Vogt-Koyanagi-Haradas disease in Southern California. Hum Immunol. 2004, 65(12):1477-82.
12. Yamamoto JH, Damico FM, Cunha-Neto E, et al. Update on autoimmune mechanisms and pathogenesis in Vogt-Koyanagi-Harada disease, a model of an autoimmune uveitis: From laboratory to reality. Módulo Temático e Simpósio Imunologia da cornea. Universidade de São Paulo. São Paulo: 2005.
13. Gocho K, Kondo I, Yamaki K, et al. Identification of specific antigen for Vogt-Koyanagi-Harada disease: lymphocyte reactivity against the tyrosinase family proteins. Invest Ophthalmol Vis Sci. 1999, 40:204.
14. Lindley DM, Boosinger TR, Cox NR. Ocular histopathology of Vogt-Koyanagi-Harada- like syndrome in an Akita dog. Vet Pathol. 1990, 27:294-6.
15. Lucena DR, Paula JS, Silva GCM, et al. Síndrome de Vogt-Koyanagi-Harada incompleta associada a HLA DRB1*01 em criança de quatro anos de idade: relato de caso. Arq Bras Oftalmol. 2007, 70(2):340-2.
16. Minoda H, Sakai J, Sugiura M, et al. High inducibility of Epstein-Barr virus replication in B lymphocytes in Vogt-Koyanagi-Harada disease. Nippon Ganka Gakkai Zasshi. 1999, 103:289-96.
17. Inomata H. Vogt-Koyanagi-Harada disease. In: Handbook of Clinical Neurology. 12a ed. São Paulo: Ed. Elsevier Science Pub; 1989.
18. Read RW, Rao NA, Cunningham ET. Vogt-Koyanagi-Harada disease. Current Opin in Ophthalmol. 2000, 11:437-42.
19. Werneck ALC, Gurgel LCA, Mello L. Sudden sensorineural hearing loss: a case report supporting the immunologic theory. Arq Neuro Psiquiatr. 2003, 61(4):1018-22.
20. Read RW, Rechodouni AK, Butani N, et al. Complications and prognostic factors in Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol. 2000.
21. Suzuki S. Quantitative evaluation of 'sunset glow' fundus in Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol. 1999, 43:327-33.
22. Walton RC. Vogt-Koyanagi-Harada Disease. Disponível em: http://www.emedicine.com/oph/topic459.htm. Acessado em: 17 de novembro de 2007.
23. Ganança MM, Caovilla HH, Munhoz MSL, et al. Alterações da audição e do equilíbrio corporal no idoso - Como Diagnosticar e Tratar. Disponível em http://www.jonas.com.br/informacao.php?info=Idoso&lg=pt. Acessado em 19 de novembro de 2007.
24. Kimura H, Ohashi N, Aso S, et al. Clinical study of the role of melanocytes in the inner ear of patients with Haradas disease. ORL J Otolaryngol Relat Spec. 1996, 58(4):233-7.
25. Amsallem P, Andrieu JG, Dehesdin D. Deafness and autoimmunity. Ann Otolaryngol Chir Cervicofac. 1985, 102(5):345-50.
26. Brito AE, Zagui RB, Rivitti EA, et al. Você conhece esta síndrome? An Bras Dermatol. 2005, 80(3):297-8.
27. Ohno S, Minakawas R, Matsuda H. Clinical studies of Vogt-Koyanagi-Haradas disease. Jpn J Ophthalmol. 1988, 32:334-343.
28. Ondrey FG, Moldestad E, Mastroianni MA, et al. Sensorineural hearing loss in Vogt-Koyanagi-Harada syndrome. Laryngoscope. 2006, 116(10):1873-6.
29. Bezerra HL, Santos LP, Carvalho AM, et al. Síndrome de Vogt-Koyanagi-Harada: revisão de 89 casos. Arq Bras Oftalmol. 1998, 61(3):331-4.
30. Sheu SJ, Kou HK, Chen JF. Original Article Prognostic Factors for Vogt-Koyanagi-Harada Disease. J Chin Med Assoc. 2003, 66:148-54.
31. Rao NA, Sukavatcharin S, Tsai JH. Vogt-Koyanagi-Harada disease diagnostic criteria. Int Ophthalmol. 2007, 27(2-3):195-9.
32. Sheu SJ. Update on uveomeningoencephalitides. Current Opinion in Neurology. 2005, 18:323-329.
33. Yawata N, Nakamura S, Kijima M, et al. High incidence of glucose intolerance in Vogt-Koyanagi-Harada disease. Br J Ophthalmol. 1999, 83:39-42.
34. Alaoui FZ, Benamour S, El Kabli H, et al. Vogt-Koyanagi-Harada syndrome: Eight cases. Rev Med Interne. 2007, 28(4):250-4.
35. Aumond MD, Leonhardt FD, Abreu CEC, et al. Síndrome de Cogan: apresentação de caso e diagnóstico diferencial. Rev Bras Otorrinolaringol. 2000, 68(3).
36. Lopis MD, Navea A, Peris C, et al. Síndrome de Vogt-Koyanagi-Harada. Actualización. Disponível em http://www.oftalmo.com/studium/studium2004/stud04-3/04c-03.htm. Acessado em 19 de novembro de 2007.
37. Andrade REA, Muccioli C, Farah ME. Injeção intravítrea de acetato de triancinolona no tratamento da síndrome de Vogt-Koyanagi-Harada. Arq Bras Oftalmol. 2004, 67(3):4001-6.
38. Berker N, Ozdamar Y, Soykan E, et al. Vogt-Koyanagi-Harada syndrome in children: report of a case and review of the literature. Ocul Immunol Inflamm. 2007, 15(4):351-7.
39. Diaz ML, Amselem L, Romero FJ, et al. Adalimumab therapy for Vogt-Koyanagi-Harada syndrome. Arch Soc Esp Oftalmol. 2007, 82(3):131-2.
40. Chan CC, Roberge FG, Whitcup SM, et al. 32 Cases of Sympathetic ophthalmia. Arch Ophthalmol. 1995, 113:597-600.
1. 5th year student of Medicine at Universidade do Estado do Pará (Monitor of Ophthalmology discipline).
2. 5th year student of Medicine at Universidade do Estado do Pará (Monitor of Otorhinolaryngology discipline).
3. Assistant teacher of ENT discipline at Universidade do Estado do Pará (Master degree in ENT by Universidade Federal do Rio de Janeiro - Doctorate student on Neuroscience by Universidade do Estado do Pará).
4. Assistant teacher of ENT discipline at Universidade do Estado do Pará and Universidade Federal do Pará (Máster degree in ENT by Universidade Federal do Rio de Janeiro - Doctorate student on Neuroscience by Universidade do Estado do Pará).
5. Adjunt Professor and Head of ENT Discipline of Universidade do Estado do Pará. Visiting Professor of ENT at Universidade Federal do Pará. Master and PhD in ENT by UNIFESP).
6. Medical school student (3rd year) at Universidade do Estado do Pará.
Institution: Universidade do Estado do Pará - UEPA (State Universtity). Belém / PA - Brazil.
Mail address:
Stephanie Gonçalves Carneiro
Travessa Rui Barbosa 619, Apto. 1301
Belém / PA - Brazil - Zip code: 66053-260
Telephone: (+55 91) 3242-7178 / 8123-1638
E-mail: stephaniecarneiro@terra.com.br
Article received on December 1st, 2007.
Article approved on March 16th, 2007.