INTRODUCTION The adrenoleukodystrophy (ALD) is a genetic disease with an X-linked heritage standard, that consists of an alteration of the peroxyssomes metabolism, and causes an accumulation of very large chain fatty acids (VLCFA) composed by 24 and 26 carbon atoms in the organism, specially in the brain and adrenal glands. Such accumulation is associated to the axons demyelinization and affects the transmission of nervous impulses and the adrenal insufficiency (1). It affects almost exclusively the male sex with beginning of symptoms between 4 and 10 years and the estimate incidence is of 1:25000 men. The clinical manifestation of the disease initially consists of alterations of behavior, hearing, vision, speech, writing, memory, gait, adrenal disorders and in the most advanced cases it occurs with generalized hypertension, loss of the cognitive, motor functions, convulsions and dysphagia. The diagnosis is confirmed by dosing the plasmatic levels of the VLCFAs, Magnetic Resonance showing demyelinizing lesions with distribution in "butterfly-wing" at the white bi-hemispheric parieto-occipital substance, electromyography compatible with myelinopathic type polyneuropathy, laboratorial research for adrenal insufficiency and karyotype whose defective gene is ABCD1 located in locus X9-28 of chromosome X (2, 3 ,4,5,6).
CASE REPORT A.V.F., 7 years old, sent to the otorhinolaryngology service for school and communication difficulties of 3 months, for auditory evaluation. The physical exam was normal and there was no precedence of auditory pathology. Audiometry was initially requested and it was carried out with a lot of difficulty with need for phonoaudiologists to guide the patient by means of writing. A bilateral moderate sensorioneural hearing loss and tympanometry with type A curve. The vocal audiometry was not performed because the child did not understand the speech at any intensity. BERA was then requested, which was normal with thresholds in 15 dB. Some months later he evolved with progressive vision loss, writing worsening and aggressiveness, and was then forwarded to the neuropediatrician with the hypothesis of neurodegenerative disease. Magnetic resonance was carried out and confirmed demyelinizing lesions in a parieto-occipital white substance. His diagnosis was confirmed through a karyotype carried out by the geneticist with immediate beginning of treatment with Lorenzo's Oil. Approximately 1 year after the beginning of the symptoms he presented with a case of choking and loss of the roaming capacity and was restricted to the wheelchair. Since then he started follow up at our dysphagia service, where the accomplishment of deglutition videoendoscopy was not possible due to the patient's intense crying and agitation; then we carried out videodeglutogram exam that confirmed oral preparatory phase oropharyngeal dysphagia and adapted pharyngeal dysphagia and in a second exam (4 moths later) he presented with severe oropharyngeal dysphagia with silent bronchoaspiration. He was forwarded to pediatric surgery for performance of gastrostomy. Today, the patient is clinically stable. The second son of the patient's family was submitted to genetic study and is not bearer of the ALD gene.
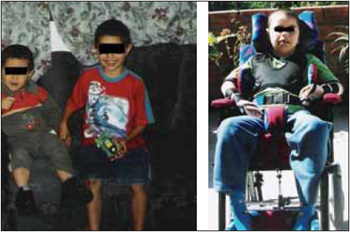
Picture 1. Pictures of the patient healthy and sick. At the top of the picture, the patient healthy and at the bottom, 1 year after the beginning of the symptoms.
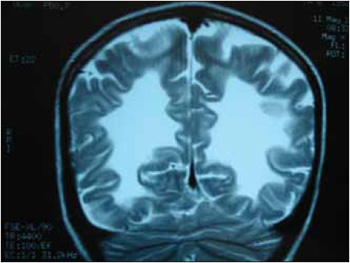
Picture 2. Coronal cut RNM. We can see the image in butterfly wing in both hemispheres.
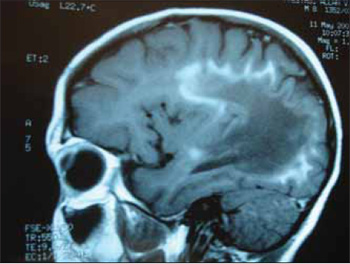
Picture 3. Sagital cut RNM. Image in butterfly wing (characteristic of the disease).
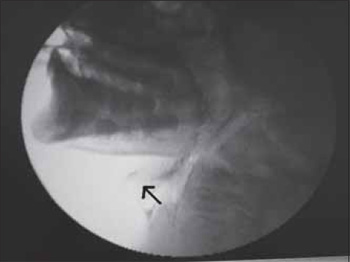
Picture 4. Deglutition videoendoscopy. Carried out one year after the beginning of the symptoms, where we observe a posterior escape of the bolus indicated by the arrow.
ALD is an uncommon genetic disease included in the group of the leukodystrophies that affects the X-linked chromosome, and is a heritage bound to the recessive sex transmitted by bearer women and fundamentally affects men. The defective gene is responsible for the encoding of an enzyme called fatty acyl CoA ligase that is found in the peroxyssomes membrane and is related to the transport of fatty acids into this cellular structure. The defective gene causes a mutation to this enzyme, the VLCFAs remain prevented from getting into the peroxyssomes and accumulate inside the cells. The precise mechanisms by means of which the VLCFAs cause a destruction in the myelin sheath are still unknown. The descent possibilities from a woman bearing ALD are of 25% of chance of having a normal male child; 25% of having an affected male child; 25% of having a normal female child; 25% of having a heterozygous female bearer child. The descent chances of an affected man, if he has daughters, is that they will all be bearers of the disease; if he has sons, they will all be normal; (1). The clinics of the disease is very variable, but the most frequent initial manifestation is the change of behavior of the child who presents with social isolation and attention deficit. In these cases the patient's hearing is usually questioned, and it may range from normal to variable grades of neurosensorial losses that are better verified with BERA, since the patient may present some cognitive prejudice, which impairs the conventional audiometry performance. When altered, BERA shows a reduction of amplitude and from the time of conduction of the waves, by suggesting a retrocochlear pathology and several thresholds alterations.
Specially in our patient, BERA was normal, with a tonal audiometry difficult to be made, which confirmed moderate neurosensorial hearing loss. At this moment, it could be a central hearing loss or a neurologic or psychosomatic disease, such as autism, for instance. The auditory potentials of medium to long latency, such as P300, could probably help us in the detection of cerebral cortex level alterations.
Other signs, such as, dysarthria, dysmetria, bradylalia, dysgraphia, graphic dyslogia or gait alterations will indicate the hypothesis of neurodegenerative disease that must be researched and diagnosed. In ALD, the diagnosis is helped by Magnetic Resonance that shows demyelinizing lesions with distribution in butterfly wing in the bihemispheric parieto-occipital white substance, dosage of the plasmatic levels of the increased VLCFAs, electromyography compatible with myelinopathic type polyneuropathy for adrenal insufficiency and karyotype. After the diagnosis a multiprofessional team is important for follow up and rehabilitation of these patients, since it is an evolutive disease. In more advanced phases they present with variable degrees of neurogenic dysphagia diagnosed through deglutition videoendoscopy or videodeglutogram. The therapy in these stages consists of deglutition or gastrostomy maneuvers where oral feeding is not safe or suitable.
There is no defined therapy for ALD so far, and according to studies, the VLCFA-free diet, such as spinach, cheese and red meat, associated to olive oil or "Lorenzo's oil" has been successful specially when managed in the beginning or before the appearing of the symptoms. The treatment of adrenal disorder, through the administration of hormones and currently the marrow transplants are treatment modalities adopted in the ALD with a very variable level of success in the disease evolution, according to the literature. Many times, the long period of time between the beginning of the symptoms and the diagnosis complicates the treatment and indicates a poor knowledge of the doctors about the subject (7,8,9,10). Specially the otorhinolaryngologist must know this disease, because he/ she is usually one of the first professionals to be sought, they may then contribute for an early diagnosis and forward them to the neuropediatrician and geneticist, and thus favor not only the patient, but also their family in the identification of bearers and genetic advisory.
FINAL COMMENTS The ALD is an uncommon neurodegenerative disease that requires early diagnosis for a better prognosis. There is no definitive therapy, but the treatment with Lorenzo's Oil has been successful associated to a multiprofessional follow up. The otorhinolaryngologist is usually one of the first professionals to be in contact with these patients, and he/she must forward them as soon as possible for diagnosis and follow them up until the more advanced phases where dysphagia is present.
BIBLIOGRAPHIC REFERENCES 1. Vargas CR, Barschak AG, Coelho DM, Furlanetto V, Souza CFM, Karam SM et col. Clinical and biochemical findings in 7 patients with X-linked adrenoleukodystrophy treated with Lorenzo's Oil. Genet. Mol. Biol. 2000, 23(4):697-701.
2. Marques PR, Melo RJV, Barraquer BL. Adrenoleucodistrofia :estudo clínico e histopatológico de um caso associado ao uso de abortivos no segundo mês de gestação. Arq. Neuropsiquiatr. 1992, 50(2):219-24.
3. Vargas CR, Barschak AG, Coelho DM, Souza CFM, Puga AS, Schwartz IVD et col. X-linked adrenoleucodystrophy: clinical and laboratory findings in 15 Brazilian patients. 2000, 23(2):261-4.
4. Echeverri PO, Espinosa EE, Moser WH, Peña SO, Barrera AL. Adrenoleucodistrofia ligada al X en ocho casos colombianos. Acta Neurol Colomb. 2005, 21(4):299-305.
5. Romero C, Martínez A, Meli F, Salas E. Desmielinización en alas de mariposa: hallazgo característico de adrenoleucodistrofia en resonancia magnética. Rev argen Radiol. 1995, 59(3):151-6.
6. Sanchez A, Parma R, Echecury M, Vazquez N, Boland R, Drittanti L, Saccoliti M, Taratuto AL. Paraplejia espástica e insuficiencia suprarrenal primaria: un caso de adrenomieloneuropatia / Spastic paraplegia and primary adrenal insuficiency: a case of adrenomyeloneuropathy. Medicina (B. Aires). 1998, 48(3):290-6.
7. Susuki Y, Imamura A, Shimozawa N, Kondo N. The clinical course of chieldhood and adolescent adrenoleukodystrophy before and after Lorenzo's oil. Brain Dev. 2001, 23(1):30-3.
8. Moser HW. Komrower Lecture. Adrenoleukodystrophy: natural history, treatment and outcomes. J. Inherit Metab Dis. 1995, 18(4):435-47.
9. Moser HW. Follow-up of 89 asyntomatic patients with adrenoleukodystrophy treated with Lorenzo's oil. Arch. Neurol. 2005, 62(7):1045-80.
10. Moser H, Dubey P, Fatemi A. Progress in X-linked adrenoleukodystrophy. Curr Opin Neurol. 2004, 17(3):263
1. Third Year Resident Doctor at the Medical School of ABC.
2. Master and Doctoral Degree at Unifesp. Assistant Professor of the Medical School of ABC.
3. Otorhinolaryngologist. Assistant Doctor of the Medical School of ABC.
4. Phonoaudiologist Especialist in Oral Motricity l, Master's Degree in Health Sciences. Phonoaudiologist at Hospital Estadual Mário Covas.
5. Doctoral Degree at the Medical School of USP. Head Professor of the Otorhinolaryngologist Discipline of the Medical School of ABC.
Institution: Faculdade de Medicina do ABC. Santo André / SP - Brazil. Mail Address: Maria Carolina Souza Queiroz - Rua Oliveira Alves, 495 - Apto. 82 - São Paulo / SP - Brazil - Zip Code: 04210-061 - Telephone: (+55 11) 2273-8131 - E-mail: carolorlabc@yahoo.com.br
Article received on March 21, 2008. Article accepted on May 27, 2009.