INTRODUÇÃOA Síndrome de Richner-Hanhart ou Tirosinemia tipo II se caracteriza por ausência em grau variável de porções distais de uma ou mais extremidades, associada à micrognasia e microglossia severa ou ambas (1).
A etiologia desta rara síndrome permanece desconhecida e, fatores ambientais, bem como genéticos têm sido postulados (2). A prevalência estimada é de 1:500.000 (1).
A patogênese do dano tecidual não é bem compreendida. Altos níveis plasmáticos de tirosina e de metabólitos desta (ácidos fenólicos) têm sido implicados, mas essa associação ainda não é confirmada (3).
Como diagnósticos diferenciais para esta síndrome, deve-se considerar a ectrodactilia, as paralisias de nervos cranianos isoladas e a Síndrome de Poland (1).
O tratamento dietético para esta patologia resulta na correção das anormalidades químicas (tirosinemias) e em acentuada resolução das lesões cutâneas e oculares. Pode, ainda, prevenir ou melhorar a afecção renal e do crescimento, mas não tem a capacidade de impedir a progressão do acometimento hepático (1).
REVISÃO DA LITERATURAA síndrome de Richner-Hanhart ou Tirosinemia tipo II (Tirosinemia Oculocutânea) é uma rara desordem autossômica recessiva ligada ao metabolismo da Tirosina, caracterizada pela deficiência da fração citosólica da Tirosina Amino Transferase Hepática (TAT). O gene responsável se encontra no cromossomo 16q22.1-q 22.3. Menos de 100 casos foram documentados, a maioria deles observada em Italianos (3).
O primeiro caso de agenesia congênita de língua foi descrito em 1718 por DE JUSSIEU, em uma menina de 15 anos. KETTNER, em 1907, foi o primeiro a associar a aglossia às severas anomalias em pés e mãos em uma criança de 4 anos. E, após pouco menos de 70 anos, em 1975, já havia quase 20 casos apresentados na literatura mundial (1).
A associação de ceratoderma palmoplantar puntiforme, ceratite e retardo mental foi primeiramente descrito, e de forma independente, por RICHNER (1938) e HANHART (1947), mas a relação com o metabolismo de tirosina não foi descoberto antes de 1973 (3).
A etiologia dessa rara síndrome permanece desconhecida e fatores ambientais e genéticos são pesquisados (2).
Na Síndrome de Richner-Hanhart, a gravidade do quadro varia de acordo com a importância das diferentes extremidades acometidas, desde peromelia completa até a ausência distal de um dedo, sindactilia ou hipoplasia ungueal. A micrognasia e a icroglossia podem ser severas e estar associadas com fissura palatina, sinéquias, signasia e oligodontia. Pode haver paralisia congênita de nervos cranianos uni ou bilateral. Habitualmente, a capacidade intelectual é preservada (1).
Há 3 tipos de tirosinemia e a apresentação dos defeitos clínico e bioquímico é diferente entre eles (3).
No tipo 1 há severo envolvimento do fígado, rins e sistema nervoso central (3). Está associada com déficit de crescimento, raquitismo, Síndrome de Fanconi, falência hepática progressiva, níveis elevados de tirosina e metionina no sangue, além de aumento na excreção urinária de tirosina e metabólitos (tirosúria) e de ácido gama-aminolevulínico. Deficiência na atividade de enzimas hepáticas, entre elas TAT e 4-hidroxipenilpiruvato dioxigenase tem sido descrita. Recentemente, a deficiência de fumarilacetoacetato hidrolase foi demonstrada em inúmeros pacientes e apresentada como o defeito enzimático primário encontrado nessa doença. São necessários NTBC e transplante hepático para o tratamento deste tipo.
A Tirosinemia tipo 2, descrita em aproximados 20 pacientes, a maioria deles com manifestações da Síndrome de Richner-Hanhart, associada à persistência de ceratite severa e hiperceratoses mais variadas em dedos e palmas das mãos e plantas dos pés, apresenta resposta completa à dieta com restrição de tirosina. A tirosina sanguínea encontra-se muito elevada e há tirosúria maciça. A ausência de atividade de TAT citosólica hepática foi demonstrada em 2 pacientes e uma deficiência intermediária foi evidenciada em 2 outros pacientes, em fígado e fibroblastos respectivamente (3).
O tipo 3 é extremamente raro, com predomínio de envolvimento neurológico (3).
RELATO DE CASOLLSS, masculino, 1 ano e 11 meses, solteiro, pardo, natural de Itaperuna / RJ e residente em São José de Ubá / RJ. Ao nascer, o pediatra observou que o recém nascido (RN) não apresentava reflexo de sucção satisfatório e que apenas conseguia mamar através da mamadeira. Por essa razão, optou por encaminhar o mesmo ao otorrinolaringologista. À oroscopia, observou-se que o RN apresentava agenesia de terço anterior da língua, associada à micrognatia; malformações dos pés, das mãos, dos pododáctilos e quirodáctilos (Figuras 1, 2, 3 e 4); desvio de septo nasal para a esquerda e ausência de adenoide. A laringe encontrava-se sem anormalidades.
Foram solicitadas radiografias das mãos e dos pés, que evidenciaram fusão dos ossos dos mesmos.
Ao analisar todas as características clínicas apresentadas pelo RN do caso relatado, concluiu-se que este era portador de uma rara síndrome, denominada Síndrome de Richner-Hanhart.
No intuito de entender a provável etiologia desta síndrome, foi interrogada à mãe a possibilidade de casamento co-sanguíneo e/ou de uso de drogas ilícitas durante a gestação, ambas negadas pela mesma. Portanto, o RN foi encaminhado ao geneticista para elucidação da causa de sua síndrome.
Atualmente, a criança é acompanhada pelo otorrinolaringologista submetida a tratamento fisioterápico. Apresenta fala apropriada e desenvolvimento motor dentro dos padrões de normalidade para idade.
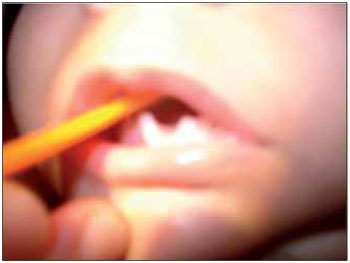
Figura 1. Imagem mostrando agenesia do terço anterior da língua.
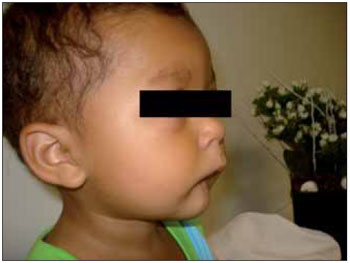
Figura 2. Imagem evidenciando a micrognatia.
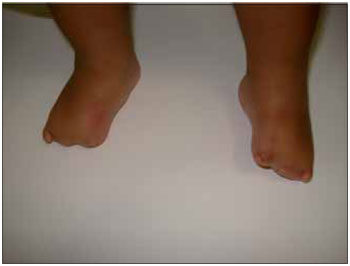
Figura 3. Imagem demonstrando anormalidade do pododáctilos.
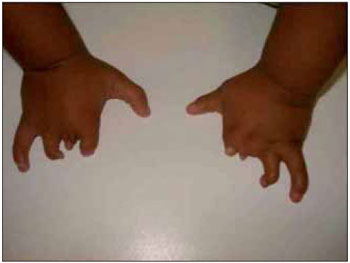
Figura 4. Imagem evidenciando anormalidade dos quirodáctilos.
A etiologia desta síndrome é desconhecida. Não se têm encontrado evidências de herança familiar e os fatores hereditários seriam pouco importantes. Alguns autores defendem a possibilidade de essa patologia ocorrer por herança genética autossômica recessiva, co-sanguinidade ou herança autossômica dominante. Outros, no entanto, consideram fatores ambientais como possíveis causadores da mesma (1).
Esta condição geralmente se apresenta na infância, com lesões cutâneas (85% dos casos), sintomas oculares (75%) ou complicações neurológicas (60%) ou qualquer combinação das três (3).
As manifestações oculares geralmente se iniciam em torno da 2ª semana de vida, enquanto que as de pele ocorrem depois de um ano de idade, mas em alguns casos pode se dá antes do 1º mês de vida (3).
Devido a micrognatia alterações no padrão de sucção podem ocorrer e necessitam de intervenção precoce, pois podem refletir nas funções de mastigação e deglutição (8). Devido ao envolvimento da língua além da mastigação, a fala pode também estar comprometida (9).
Dieta baixa em tirosina e fenilananina, em combinação com a administração de vitamina C, reduzem os níveis séricos de tirosina. No entanto, em alguns pacientes não é possível impedir o progresso da enfermidade somente com a dieta e, portanto, se torna necessário o transplante hepático (6).
COMENTÁRIOS FINAISA Síndrome de Richner-Hanhart é extremamente infrequente. Entretanto, se torna importante descrevê-la, a fim de saber reconhecê-la e diagnosticá-la precocemente e, ainda, prosseguir com estudos que norteiam o esclarecimento de suas prováveis etiologias, sejam elas hereditárias ou ambientais.
REFERÊNCIAS BIBLIOGRÁFICAS1. Castillo ST, Rojas JZ, Monasterio LA. Sindrome de Hanhart. Rev. Chil. Pediatr. 1985, 56(3):180-183.
2. Gillerot Y, Maldergem LV, Chef R, Koulischer L. Hypoglossia-hypodactyly syndrome with hydrocephalus: a clue to the aetiology? J Med Genet. 1991, 28: 490-491.
3. Paige DG, Clayton P, Bowron A, Harper JI. Richner-Hanhart syndrome (oculo cutaneoustyrosinaemia, tyrosinaemia type II). Journal of the Royal Society of Medicine. 1992, 85:759-760 .
4. Janakiraman L, Sathiyasekaran M, Deenadayalan M, Ganesh R, Mahesh U. Richner Hanhart Syndrome. Indian Journal of Pediatrics. 2006, 73:161-162.
5. Ramos BE, Pascual MMJ. Tratamiento dietético de las enfermedades metabólicas. Informacion terapeutica del sistema nacional de salud. 2005, 29(4):81-95.
6. Monroy LC, Urbanes MB, Sedan CA. Tirosinosis: Reporte de un caso. Revista Colombiana de Padiatría.
7. Cantani GO, Kennaway A, D'eufemia NGP. Tirosinemia crônica associada com deficiência de 41-hidroxifenilpiruvato dioxigenase com ataxia aguda intermitente e sem envolvimento visceral e ósseo. Pediatr. Res. 1983, 17:25-29.
8. Silva RCL, Alves FFS, Netto SSG, Silva CM. As alterações fonoaudiológicas na síndrome de Goldenhar - relato de caso. Rev Soc Bras Fonoaudiol. 2008, 13(3):290-5.
9. Freitas AC, Filho PN, Queiroz AM, Assed S, Silva FWGP. Síndrome de Moebius: Relato de caso clínico. Revista de Odontologia da Universidade Cidade de São Paulo. 2006, 18(3):297-302.
1) Especialista em Otorrinolaringologia no Hospital das Clínicas em Itaperuna.
2) Estagiária de Otorrinolaringologia. Acadêmica do sexto ano de Medicina.
3) Residente de Otorrinolaringologia do Hospital São José do Avaí / RJ.
4) Acadêmica do Sexto Ano de Medicina.
5) Estagiário de Cirurgia Geral do Hospital São José do Avaí. Acadêmica do sexto ano de Medicina.
Instituição: Hospital das Clínicas. Itaperuna / RJ - Brasil. Endereço para correspondência: Daniela Silva Pais - Rua Rui Barbosa 383, 602 - Itaperuna / RJ - Brasil - CEP: 28300-000 - Telefone: (+55 22) 8801-5160 - E-mail: danielasilvapais@gmail.com
Artigo recebido em 24 de Agosto de 2009. Artigo aprovado em 27 de Janeiro de 2010.